Characterization of hydrocarbon degrading microorganisms from Glycine max and Zea mays phytoremediated crude oil contaminated soil
Article information

Abstract
Microbe-plant partnership in phytoremediation involves a synergistic interaction that leads to degradation of contaminants. The identification and characterization of these microorganisms is fundamental in environmental management. This study is aimed at investigating the influence of Glycine max and Zea mays on microbial make-up and differentiation of soil bacterial and fungal isolates in crude oil contaminated soil. We employed conventional technique of microbial isolation and gene sequencing to evaluate the microbial composition in crude oil contaminated soil. The microorganisms were isolated from crude oil contaminated soil (0%, 4%, 8%) and were identified using 16S rRNA gene (for bacteria) and Internal Transcribed Spacer (ITS) gene (for fungi). We observed a change in the microbial cell density with respect to treatment conditions implying a shift in microbial dynamics to total hydrocarbon utilizing bacteria as the dominant microbes. The sequence data revealed five bacteria strain; Klebsiella aerogenes strain 77, Klebsiella aerogenes strain UISO178, Salmonella enterica strain ABUH7, Klebsiella aerogenes strain M242 and Enterobacter sp. NCCP-607 and three fungi strains; Galactomyces geotrichum strain CBS, Aspergillus niger strain YMCHA73 and Trichoderma virens isolate A701. Annotation analysis using FGENESB and gene scan revealed proteins involved in various metabolic processes and hydrocarbon utilization. GHOSTKOLA output revealed several genetic elements and pathways such as DnaA, PYG, mrcA, environmental, cellular and genetic information processing and degradation enhancers. Our findings show that G. max and Z. mays in association with bacteria can enhance ecosystem restoration of crude oil contaminated soil.
Introduction
Crude oil contamination and pollution are inevitable in oil producing regions [1] that use it as its major source of revenue. The chemical composition of crude oil can be harmful to the living system [2] hence, the removal of this contamination is essential to saving the living organisms in such polluted area [1]. As an alternative to physical and chemical methods, phytoremediation has been a promising approach to clean up contaminated soils [3,4].
The combined action of plants and microbes (the rhizosphere-mediated bioremediation) [5] is an important component of rhizosphere ecology. In this synergistic interaction, the plants support the growth of microbes by improving soil condition through root activities and release of root exudate [6]. The microbes play an important role in contaminant degradation, thus, reducing phytotoxicity and allowing the plants to grow in the environment [7]. Moreover, root exudates provide source of energy and nutrients to microbes thereby enhancing microbial activities, and in turn, microbes stimulate plant exudation from plant roots. Plants-microbes interaction allows plants and their associated microbes to coexist or support each other for survival in the changing environment. The outcomes of these interactions are as follows: elevated number of organisms, change in diversity/metabolic activity of the microbes able to degrade contaminants and stimulation of plant growth. In some cases, a reduced activity, metabolism and cell number are inevitable. In petroleum hydrocarbon environment, mycorrhizae (the close and generally mutualistic relationships between plants and fungi) have been identified as ‘tripartite associations’, as they are influenced by the interactive activities of plants, fungi and bacteria [8]. The symbiotic association between mycorrhizal fungi, plants and bacteria can suggest the hierarchical trajectory in a broader range of niches and their role in the rhizosphere. Soil-inhabiting fungal and bacterial strains are often grouped together under the broad term ‘microbial community’, but these groups may not respond similarly to either hydrocarbon contaminants or plant introduction because of differences in physiology and ecology. However, these interactions are important in hydrocarbon degradation because their co-participation enhances hydrocarbon degradation. Plant-microbe partnership is a promising strategy for the remediation of soil and water polluted with hydrocarbons [9].
Various microbes have been reported by different scholars to colonize the rhizosphere or endosphere of plants involved in the degradation of petroleum hydrocarbons [10–12]. However, a lot of microbes in the rhizosphere of the soil are yet to be identified thus the need for their identification and their roles. Furthermore, the genetic characteristics, the interaction between beneficial microbes and plants such as Z. mays and G. max as well as the environment of hydrocarbon degrading genes have not been well characterized. Successful implementation of phytoremediation will depend on many parameters including the complexity of the competing microflora, the presence of a compatible plant species, microbial species-dependent capability (such as presence of catabolic genes encoding degradation enzymes and induction of stress proteins in order to maintain activity and survival of the microbe) and other environmental factors, which might enhance or decrease the survival of a particular strain. Efficient rhizosphere colonization by microbes is likely to enhance plants’ survival and metabolic activities.
The aim of this study was to characterize hydrocarbon degrading microorganisms from crude oil contaminated soil remediated with Z. mays and G. max. Here, we used 16s rRNA and ITS to identify bacteria and fungi isolated from rhizosphere of crude oil contaminated soil after remediation with Z. mays and G. max. The basis of their selection is that among other plants investigated, Z. mays and G. max showed better physiological attributes which include having excellent ability to remove hydrocarbons [13,14]. Therefore, we hypothesize that the microbial strains in association with plants can have beneficial roles towards physiological characteristics. Hence, there is a need to characterize remediated soil to identify degrading microflora with high hydrocarbon degradation potential, which also efficiently colonize the environment (e.g. the rhizosphere) of an appropriate plant species used for phytoremediation.
Materials and Methods
Sample collection and preparation
Soil samples were collected from the root zones of G. max and Z. mays used for phytoremediation studies and untreated contaminated soil was used as control. The plants were grown in a pot experiment for four months. The soil samples were labelled as follows: soil remediated with G. max, soil remediated with Z. mays, soil remediated with G. max and Z. mays, soil remediated with Z. mays fortified with poultry dung, soil remediated with G. max fortified with poultry dung, soil remediated with G. max and Z. mays fortified with poultry dung, soil only and soil with poultry dung only. The treatment application was from 0% to 8% crude oil concentration. The experiment was done in triplicates. The control (0% crude oil contamination) did not receive crude oil treatment. The soil samples were collected using a sterilized hand trowel and kept in well labelled nylon packs as described in Njoku et al. [15].
Isolation of bacteria and fungi and extraction of genomic DNA
Isolation of the bacterial and fungal isolates from the soil samples followed same protocols described in Njoku et al [15]. The microbial load was determined using the plate count method. Genomic DNA extraction was done following the protocol of Trindade et al. [16] using 50–100 mg (wet weight) fungal or bacterial cells, 200 μL lysing solution using Zymo spin kit. The quantity and quality of the extracted DNA was determined using a Nanodrop of model 2000 from Thermoscientific, measured at 260/280 nm [17].
Polymerase chain reaction analysis
The DNA was subjected to a cocktail mix consisting of 10 μL of 5 x GoTaq colourless reaction, 3 μL of MgCl2, 1 μL of 10 mM of dNTPs mix, 1 μL of 10 pmol each. Internal Transcribed Spacer (ITS) gene was used to characterize fungi and 16S rRNA was used to characterize bacteria. The purified fragment was checked on 2% agarose gel ran on a voltage of 120 V for one hour [16].
- ITS4 TCCTCCGCTTATTGACATGS
- ITS5 GGAACTAAAAGTCGTAACAAGG
- 16SF: GTGCCAGCAGCCGCGCTAA
- 16SR: AGACCCGGGAACGTATTCAC
Genomic sequencing and annotation
The amplified fragments were sequenced using a genetic analyser 3130 x1 sequencer from Applied Biosystems, Waltham, Massachusetts, USA, using manufacturer’s manual and the kit used was Big Dye terminator v3.1 cycle sequencing kit. All genetic analyses were carried out using Bio-Edit software version 7.0 as was described by Hall [18] and MEGA 6 [19]. Organism identification was done using the BLAST search against the nonredundant version of the NCBI reference database while protein-coding genes annotation was done using GENESCAN, FGENESB and GHOSTKOLA annotation pipelines [15,20].
Phylogenic analysis
Phylogenic relationship analysis was performed on the isolates’ sequences using Molecular Evolutionary Genetic Analysis version 6 [21], analysing both bacterial and fungal isolate sequences differently and then comparing them together to estimate the isolates occurrence during the study. Sequences were prepared using FASTA format and aligned using Clusta W option of the program [21].
Statistical analysis
One-way analysis of variance (ANOVA) at P<0.05 was used to test the significance of experiment followed by LSD test at P<0.05 confidence interval. All data were processed using GraphPad (Version 9.0) [15].
Results and Discussion
Bacterial and fungal isolates from crude oil contaminated soil
The microbial colony counts from soil treated with different concentrations of crude oil are shown in Figure 1 and Table S1. The hydrocarbon significantly reduced total heterotrophic bacteria. On day 1 of the experiment (immediately after crude oil treatment), the total heterotrophic bacteria count reduced from 2.1×106 cfu/g in the control experiment (without crude oil) to 1.0×106 cfu/g in 8% treatment. On day 120, such reduction was still prevalent though it was typically based on plant species and treatment type. The maximum heterotrophic counts were observed when the two plants (G. max and Z. mays) with poultry dung interacted Figure 1. This suggests that poultry dung can be a good enhancer of microbial community. The heterotrophic counts decreased from 36.0×106 cfu/g in the controlled experiment (without crude oil) to 20.0×106 cfu/g in 8% treatment given 44% reduction. In the absence of these plants, the heterotrophic counts reduced from 12.25×106 cfu/g in the control experiment (without crude oil) to 3.8×106 cfu/g in 8% treatment given 69% loss of total heterotrophic bacteria. This suggests that the two plants have mechanisms to preserve microbial biodiversity. Contrary to this trend, hydrocarbon utilising bacteria (HUB) increased in responding to crude oil treatments. For instance, contaminated soil planted with G. max recorded an increase in microbial count ranging from 1.5 ×105 cfu/g in 0% crude oil treatment to 4.2×105 cfu/g in 4% crude oil treatment and 4.9×105 cfu/g in 8% crude oil treatment after remediation. Likewise, increase in microbial count from 1.2×105 cfu/g in 0% crude oil treatment to 5.2×105 cfu/g in 8% crude oil treatment was observed after remediation with Z. mays. This shows 71% increase. When the two plants (G. max and Z. mays) with poultry dung interacted, the trend changed increasing from 2.3×105 cfu/g in 0% crude oil treatment to 6.0×105 cfu/g in 4% crude oil treatment, thereafter reduced to 5.2×105 cfu/g in 8% crude oil treatment. The contaminated soil without these plants did not show much increase in hydrocarbon utilizing bacteria. The greater increase of microbe in the contaminated soils with plants could be because root exudates provide sufficient carbon and energy to support large numbers of microbes [15].
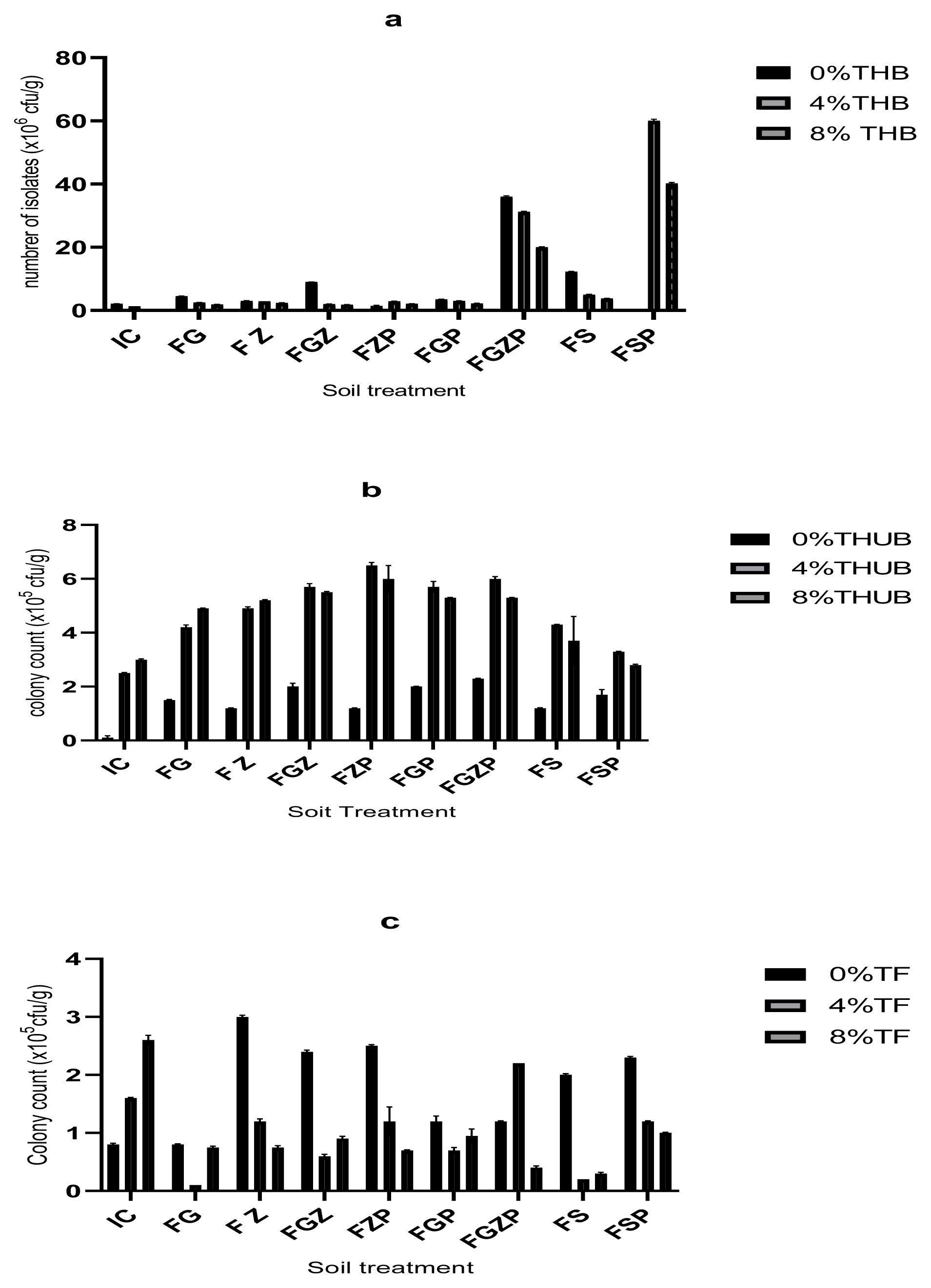
The microbial colony count from the rhizosphere soil on day 1 (initial count) and on day 120 (final count) of the study; a =total heterotrophic bacteria, b = total hydrocarbon utilizing bacteria, total fungi; IC =initial count, FG = final count+G. max, FZ = Final count+Z. mays, final count+G. max+Z. mays, FZP = Final count+Z. mays + poultry dung, FGP =Final count+G. max+poultry dung, FGZP = Final count+G. max+Z. mays+poultry dung, FS= Final count in soil alone, FSP = final count in soil+poultry dung.
Crude oil contamination caused a reductive effect on the fungal counts Table S1. The fungal counts decreased with increasing concentrations of crude oil except the soil augmented with G. max, Z. mays and poultry dung. Increase in the fungal count was observed in 4% crude oil treatment (from 1.2×105 cfu/g in control to 2.2×105 cfu/g) which decreased after phytoremediation. This could also imply the supportive function of the two plants in microbial biodiversity conservation. Highest fungal count was observed in 8% treatment after the initial day of 2.6×105 cfu/g while the lowest was found in soil with G. max, Z. mays and poultry dung with 4.0×104 cfu/g.
Plant activities are influenced by many aspects of the soil environment, including nutrient availability and the abiotic factors. It has been reported by Arslan et al. [9] that during phytoremediation of hydrocarbon contaminated soil, competition for nutrients between plants and their associated microorganisms is inevitable. Hence, nutrients act as limiting factors for plant growth and microbial proliferation. Further, Arslan et al. [9] also reported that nutrients enhanced the abundance and expression of alkane hydroxylase CYP153 gene in the rhizosphere of ryegrass planted in hydrocarbon-polluted soil. In this study, we noticed enhanced bacterial and fungal colony in the presence of poultry dung which can be substantiated by the reason given by Arslan et al. [9]. More so, we found out that introduction of the G. max and Z. mays influenced the colony counts of soil bacteria and fungi in crude oil contaminated environment. We can therefore infer a change in community structure and composition of microbes due to the growth of Z. mays and G. max which may subsequently result to a shift in community dynamics as reported by other researchers in metagenomics studies [22]. The concentration of petroleum hydrocarbon contaminants and plants species in each treatment had a strong significant effect on both bacterial and fungal colonies. It suggests that petroleum hydrocarbon contamination leads to a shift in the microbial composition to total hydrocarbon utilizing bacteria as the dominant microbes. Cultivable fungal diversity has previously been shown to decrease at high petroleum hydrocarbon concentrations [23]. This also suggests that bacteria can be a key player in hydrocarbon degradation. de Lima e Silva et al. [24] showed that high population density of total hydrocarbon utilizing bacteria indicates that the bacteria are able to use crude oil hydrocarbon as the sole source of carbon and energy thus, playing a key role in degradation processes. Previous studies have shown that plants roots stimulate rhizosphere microbial communities by aerating the soil and releasing exoenzymes as well as nutrients through root exudates. They also provide surface for colonization and niches to protect soil microbes against desiccation and other abiotic and biotic stresses [25]. These could be the reasons for higher abundances of microbes in the rhizosphere which are also observed in the current study.
This study shows that although Z. mays plays more active role in enhancing fungal and bacterial population than G. max, their combined effort is more effective in enhancing microbial load. Li et al. [26] reported that legumes increase basal respiration and metabolic quotient and have a positive effect on activity and functional diversity of the soil microbial community. Hannula et al. [27] also reported significant effect of plant groups (forbs, grasses and legumes) on the community composition of both soil fungi and bacteria. Several studies have also shown that plants have species specific effects on the diversity and structure of the soil microbial community [28,29] hence the difference in the abundance of the microbes in the soils with different plants observed in this study. We also noticed that plants alone cannot shape the microbiome ecology rather addition of certain amendments such as poultry dung can improve microbiome community structure under crude oil stress. Thus increase in bacteria population size with the addition of poultry dung indicates that it enhances microbial activities as reported in the case of cow dung by Njoku et al. [30].
The quantity and quality of DNA of the bacterial and fungal isolates
The quantity and quality of DNA from the isolates are shown in Figure 2. The DNA yield of Salmonella enterica strain ABUH7 (isolate c) was highest (222.00 ng/μL) followed that of Klebsiella aerogenes strain M242 (isolate d) with DNA yield of 131.20 ng/μL, while fungi isolate Galactomyces geotrichum (isolate f) had the lowest DNA yield (25.70 ng/μL). On the other hand, K. aerogenes strain 77 had the best DNA quality (1.8). S. enterica strain ABUH 7 had the lowest in DNA quality of 0.73 followed by K. aerogenes strain M242 (0.74). Both the quality and quantity of the DNA were not significantly different from each other at P≥0.05, 0.01 and 0.001. Accurate DNA quantity measures are critical for biological process optimization. The quantity and purity of DNA show how contaminants affect assessment performance of biological system. Purity ratios, obtained in this study were within the accepted range (1.5–2.2) suggesting absence of RNA co-extraction and PCR inhibition [31]. In this study, the quantity (concentration) and quality (purity) of the DNA extract varied by microbial type demonstrating different characteristics of the organisms. The differences in the purity indicate the reason for the variation in DNA concentration. According to Olson and Morrow [30], DNA concentration (quantity) is dependent on sample purity. This study found out that bacteria especially S. enterica strain ABUH7 yielded more than others. This can assist them on their physiological roles. The quantity of DNA may have influence on the fragment length and GC content hence, the physiological roles of the microbes. This suggests the possibility of the bacteria with the highest DNA quantity (S. enterica strain ABUH 7), to be a good candidate for bioremediation.
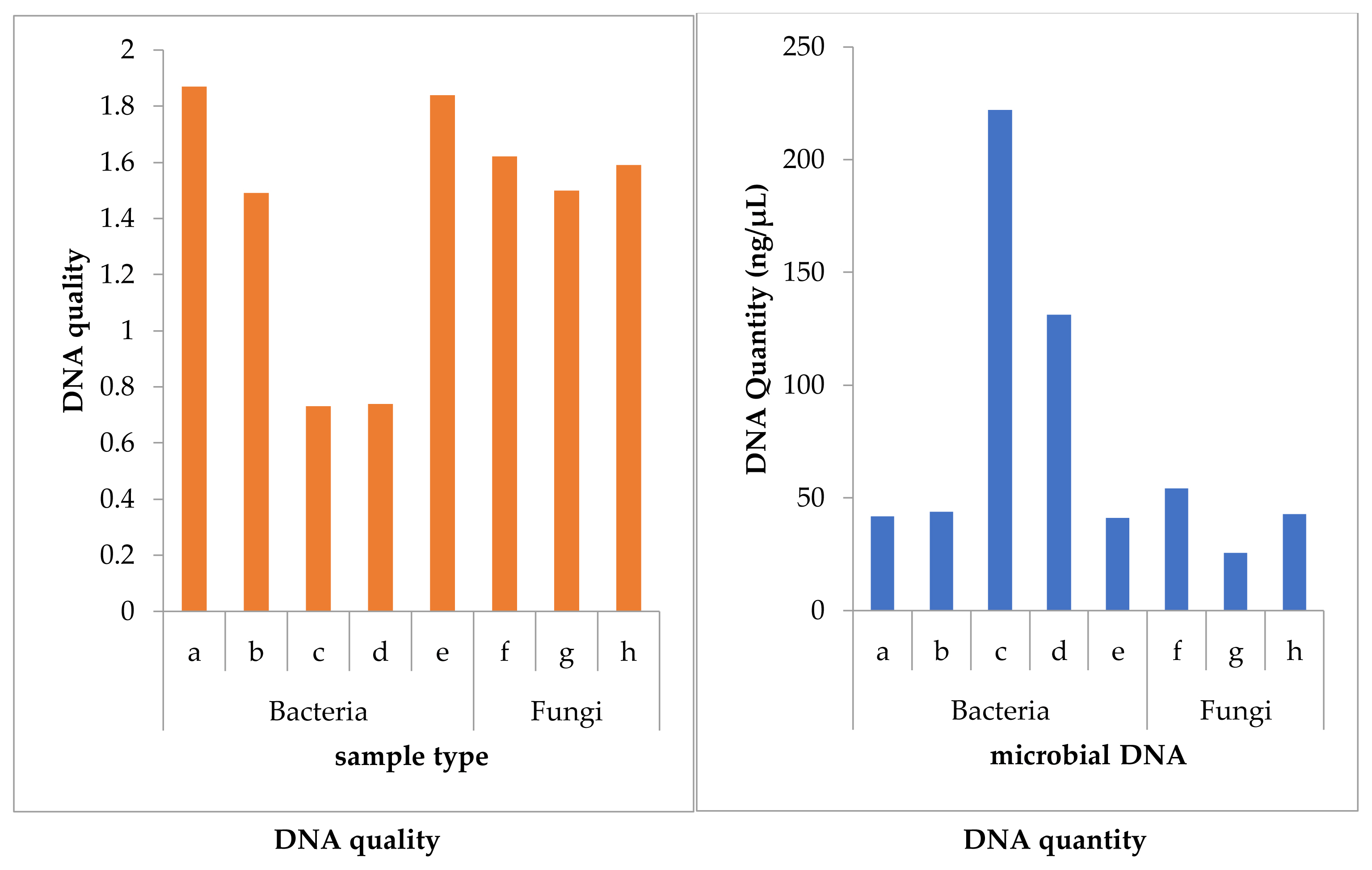
The quality and quantity of the DNA from the different isolates: a= K. aerogenes, strain 77, b = K. aerogenes strain UISO178, c= S. enterica strain ABUH 7, d= K. aerogenes strain M242, e= E. sp. NCCP-607, f= G. geotrichum CBS774.7, g= A. niger YMCHA 73, h= T. virens A701
Molecular Identity and characteristics of bacterial and fungal isolates
The molecularly identified isolates are presented in Table 1. Internal Transcribed Spacer (ITS) gene and 16S rRNA gene for characterizing fungi and bacteria revealed amplification of all the fragments. Analysis of the PCR products revealed various bands of different sizes Figure S1. Sequence analysis revealed various reads which were identified using BLAST. BLAST analysis revealed that the isolates are of eight species Table 2. Majority of the isolated bacteria belonged to Klebsiella species. The overall guanosine plus cytosine nucleotide (G+C) content of bacteria were within 54% while in fungi, it ranged from 42.54% in G. geotrichum to 57.63% in Aspergillus niger (lowest GC to highest GC respectively). The amplified purified PCR products examined in gel electrophoresis revealed a band with molecular size ~1500. More so, fungi isolates examined showed bands with molecular sizes ranged between ≤1200 bp Figure S1.

Summary of bacterial and fungal sequence characteristics

The protein-coding genes annotated with GENESCAN and FGENESB.
The greater abundance of Klebsiella species in the samples might have resulted from their high ability to tolerate and degrade aromatic hydrocarbon [32]. Hence, this suggests their ability to use hydrocarbon as energy source. Similar result was reported by Rodrigues et al. [33] that Klebsiella strains were widespread in the estuary and have the ability to degrade hydrocarbons. However, the colonization of Klebsiella strains in the rhizosphere of plants is first reported in this work though the mechanism is not yet known. S. enterica strains are another bacteria species present in the contaminated soil. Schikora et al. [34] revealed that Salmonella serovars are highly plant species specific and that different plants have various levels of resistance towards these bacteria. Therefore the presence of S. enterica in the rhizosphere of G. max and Z. mays can suggest that they can be included as plants that can tolerate them. G. geotrichum plays various roles in degradation of various hazardous substances [35]. This suggests that the microbe could have played a role in the degradation of crude oil in this study as it was observed to be present in the soils used in this study. The genus Enterobacter comprises a range of beneficial plant-associated bacteria showing plant growth promotion [36]. Therefore, we can suggest that the Enterobacter species found in our result, in association with G. max and Z. mays can have beneficial capacities towards crude oil remediation and possibly supported the growth of the plants.
A. niger has substantial potential for the biodegradation of diverse pollutants hence, its’ availability in the phytoremediated soil was not a surprise. Adongbede and Majekodunni [37] found out that A. niger co-existed with Helianthus annuus rhizosphere. Also and Njoku [13] reported A. niger to be among different microbes in crude oil contaminated soil remediated with G. max. In this research, it can be posited that A. niger can be suitable with G. max and Z. mays plants for partnership for remediation of crude oil contaminated soils. The presence of the microoorganisms identified in the soils in this can be attributed to their roles and nature. The clustering of cultivar-specific bacterial and fungi species showed differences within the treatment. We posit that the abundances of each species of bacteria and fungi may differ. As a result, one of the main roles of effective plants in phytoremediation of hydrocarbons may be in selecting fungi that are synergistic with, or at least not antagonistic to, target hydrocarbon-degrading bacteria [38].
Aside from changes in the abundance of organisms, the abiotic environment can influence the nucleotide composition [38]. High GC base pair has been shown to confer higher thermal stability and helps in tolerance to oil toxicity [16]. In bacteria, an increase in GC content correlates with a broader tolerance range for a species to environmental stress [39]. The A. niger YMCHA73 and T. virens A701 with higher GC contents which we isolated in this study can tolerate environmental stress and can be useful in bioremediation of crude oil. It is possible that they assisted in the remediation of crude oil in this study.
Phylogenic relationship of the isolated microorganisms
The phylogenic relationship of the nucleotide sequences of the organisms compared with each other is shown in Figure 3. The Neighbourhood-Joining distance analysis with sequence difference and topology showed distinct lineages belonging to the clusters of fungi and bacteria. In the fungal group, G. geotrichum was an out cluster while A. niger and T. virens were more related. In bacteria cluster, K. aerogens strain UISO178 and S. enterica strain ABUH7 were more related to each other compared to others.

Phylogenetic relationship among all microbial isolates observed in the study.
The reason for the presence of the two clusters in the remediated soil could be due to combination of selective factors, proximity and functional capacity of these microbes [15]. Phylogenetically, distant lineages can share common functional features. Most microbial sequences analyzed in different taxonomic divisions could be related to representatives with known metabolic traits. We therefore, suggest that this phylogenetic relationship may have functional implications on phytoremediation of crude oil contaminated environment.
Annotated protein coding genes
The annotated microorganism’s genomes using FGENESB and GENESCAN are shown in Table 2.
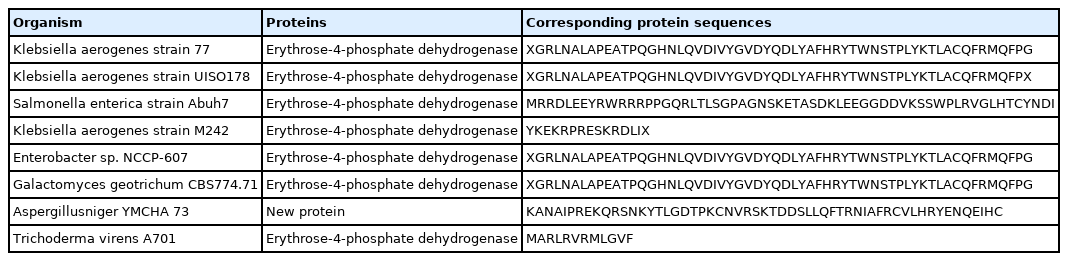
The predicted potential genes and proteins using FGENESB are shown in Table 3 while Table 4 shows the predicted potential peptides in microbial genomes using GENESCAN. Several genes and their proteins were found. Annotation using GENESCAN showed the presence of single protein coding gene sequence in all the organisms identified. All the organisms have genes coding for the enzyme Erythrose-4-phosphate dehydrogenase that functions to remove toxic substances, except for A. niger in which a new protein was identified. Dehydrogenase belongs to the group of oxidoreductases that oxidize a substrate by reducing an electron acceptor, usually NAD+/NADP+ or a flavin coenzyme such as FAD or FMN [39]. These enzymes are largely responsible for the degradation of toxic substances.

Predicted potential genes and protein in microbial gene sequence using FGENESB.
Predicted potential peptides in microbial genomes using GENESCAN.
Protein coding genes and genetic elements of the bacteria and fungi strains
Protein coding genes from the bacterial and fungal strains annotated above Table 2 were assembled and compared with GhostKOALA database to gain insight into the possible roles of the proteins Table 5. It is interesting to note that several genetic elements were annotated. In K. aerogenes, strain DAS43, DnaA responsible for cellular, environmental and genetic information processing was annotated. In Salmonella strains, pbpC and mrcA responsible for peptidoglycan biosynthesis and degradation were found. Few hydrocarbon degrading enzymes such as transferases, and hydrolases were annotated. Different variants of genes in addition to drug resistance and ATP-dependent helicase were found in K. aerogenes strain KCTC 2190. Enterobacter aerogenes strain ATCC 13048 harbours the degradative enhancer genes (EDEM1) and PYG, glgP genes. However, it was also found to have infection related genes Table S2. Careful comparisons of the gene sequence suggested the presence of set of similar genes among these organisms.

Protein coding genes and genetic elements of the bacteria and fungi strains.
The gene statistics suggested that there is intraspecies variation of genetic elements in the microbes. The genome data supported and extended various laboratory observations in the plant growth promotion attributes. Different genes that are involved in environmental, cellular, biosynthesis, degradation, enhancers and genetic information are identified. dnaA gene was found to be responsible for cellular processing (replication) in K. aerogenes, strain 77 while mrcA, pbpC genes were found to be present in Enterobacter sp. NCCP-607. DnaA plays a key role in the initiation and regulation of chromosomal replication [40]. It binds in an ATP-dependent fashion to the origin of replication (oriC) to initiate formation of the DNA replication initiation complex exactly once per cell cycle. Leonard et al. [41] found out that for E. coli, DnaA-ATP is needed for site recognition and occupation, so that initiation timing is coupled to DnaA-ATP levels. The presence of this gene can be the reason for the abundance of Klebsiella species in the hydrocarbon contaminated samples because it can help in resisting toxic substances in the environment. Furthermore, mrcA gene also plays a cellular role and contains enzymes such as transferases and hydrolases which are responsible for metabolism, biosynthesis, degradation and drug resistance. K. aerogenes strainM242 contains important genes known as EDEM whose role is associated to protein processing. They are also known to be degradative enhancers. This can be important in metabolism of xenobiotics.
Interestingly, except K aerogenes strain M242, Klebsiella species did not habour any specific infection related gene. G. geotrichum, A. niger and T. virens were found to have infection-related genes. Previously, Rajkumari et al. [42] had also reported some Klebsiella species without any infection related gene. Similarly, K. pneumoniae KP617 had been reported to harbor 117 virulence genes, but did not possess any unique virulence factors [43]. It is likely that the infectious related genes we identified in some of the organisms may not possess virulent factors supporting their colonization in plants rhizosphere without infecting the plants.
The bacteria and fungi were isolated from oil-contaminated soil in this study have potential for implementation in oilfield bioremediation. Numerous genes associated with hydrocarbon degradation were identified. The gene sets available in the genome indicate that the organisms metabolize hydrocarbons using tricarboxylic acid cycle pathways intermediates. K. pneumoniae 342 strains had been suggested to metabolize hydrocarbons in the similar pathway [44]. Identification of benzoate pathway indicates that the bacteria have the ability to degrade benzoate through ortho-cleavage of β-ketoadipate pathway forming cis–cis muconic acid (ccMA) as an intermediate of this pathway leading to tricarboxylic acid cycle (TCA) intermediates [42]. The presence of various intermediates suggests that the bacteria have a broad potential for transformation of xenobiotics.
Conclusions
Interactions between plants, bacteria and fungi occur within the rhizosphere; however, the link between these groups is altered in soils that have been disturbed by high concentrations of hydrocarbon contaminants. The study shows that fungal communities were more sensitive to hydrocarbons relative to bacteria by low enumeration of fungal counts compared to hydrocarbon utilizing bacteria. The enumeration of large hydrocarbon utilizing bacteria could be because the microorganisms use crude oil as a sole source of carbon and energy, hence suggesting their role in hydrocarbon remediation. A significant homology of the genes involved in hydrocarbon degradation was observed through the comparative analysis of the gene sequence. Dehydrogenase enzymes that function in removal of toxic substances were predicted in these sequences, suggesting a possible role for bioremediation. Actually, overlapping functional characteristics like biodegradation and virulence were identified in some sequence data. This can be exploited for the remediation of crude oil polluted sites as these microbes have diverse metabolic potential that could be exploited for additional environmental and biotechnological applications. In this study it is suggested that microbial structure and composition can be modelled and enhanced if appropriate plants are in partnership with bacteria in a stressed condition like crude oil.
Supplementary Material
Table S1.
Microbial colony count from rhizosphere soil on day 1 (initial count) and day 120 (final count) of the study.
Table S2.
Predicted infections related diseases in the bacteria and fungi strains.
Figure S1.
The gel electrophoresis of purified amplified PCR products with band sizes run on the gel for bacteria (a) and fungi (b). M=DNA Ladder; a=K. aerogenes, strain 77 band; b=K. aerogenes strain UISO178 band; c=S. enterica strain ABUH 7 band; d=K. aerogenes strain M242 band; e=E. sp. NCCP-607 band, f=G. geotrichum CBS774.71 band; g=A. niger YMCHA 73 band, h=T. virens A701 band.
Notes
The authors declare that there is no competing interest to influence the work reported in this paper.
CRediT author statement
KLN: Conceptualization, Supervision, Editing ; EOU: Investigation, Data Analyses, Writing; TOJ: Investigation, visulization, OZA: Original Draft Preparation; POI: Reviewing, Editing