Toxic detection in mine water based on proteomic analysis of lysosomal enzymes in Saccharomyces cerevisiae
Article information
Abstract
Objectives
Lysosome is the cell-organelle which is commonly used as biomonitoring tool in environmental pollution. In this study, the lysosomal proteomic of the yeast Saccharomyces cerevisiae was analyzed for utilization in the detection of toxic substances in mine water samples.
Methods
This work informs the expression of lysosomal proteomic in yeast in response with toxic chemicals, such as sodium meta-arsenite and tetracycline, for screening specific biomarkers. After that, a recombinant yeast contained this biomarker were constructed for toxic detection in pure toxic chemicals and mine water samples.
Results
Each chemical had an optimal dose at which the fluorescent protein intensity reached the peak. In the case of water samples, the yeast showed the response with sample 1, 3, 4, and 5; whereas there is no response with sample 2, 6, and 7.
Conclusions
The recombinant yeast showed a high ability of toxic detection in response with several chemicals such as heavy metals and pharmaceuticals. In the case of mine water samples, the response varied depending on the sample content.
Introduction
In recent years, there has been growing concern about the harmfulness of pollutants present in the environment. There is currently a need for sensitive, rapid, robust and cheap toxicity assay that can be used for the screening and monitoring of a wide range of toxic chemicals in the environment. Lysosomal responses are broadly used as biomarker indicative of the general stress caused by pollution in the aquatic environment. The destabilization of lysosomal membrane causing by environmental stressors results in functional alterations and the release of hydrolytic enzymes into the cytosol. The lysosomal membrane stability (LMS) has been used in field studies [1-3] and has been developed to new technique named neutral red retention assay. In addition to the LMS, the change in lysosomal dimensions has been also widely used in revealing the stress in aquatic environment, in case of mussels and fish [4,5]. However, these approaches were unstable for the onsite monitoring, time-consuming and focused only on toxic substances which damaged lysosomal membrane. Therefore, a new approach on lysosome, which had a huge potentiality in toxic detection, needs to be discovered.
In previous works, the exposure of some organisms to oxidative stresses due to toxic agents alters lysosomal enzymes, thus we used intracellular lysosomes in Saccharomyces cerevisiae as a biomonitoring tool to detect oxidative stresses by pesticides and heavy metals. Beside this, the effects of tetracycline and aspirin were evaluated simultaneously to examine non-causing oxidative stress agent influence. The lysosomes ability to detect toxicity was evaluated by using confocal microscope after staining lysosomes with LysoTracker. Treatment of S. cerevisiae with toxic substances increased the number of conspicuous and red lysosome- like organelles surrounding S. cerevisiae nucleus. The results indicated that each chemical has an optimal concentration at which the quantity of lysosomes reach the peak while the growth of yeast were not affected. It means that our method can detect the sub-lethal concentrations of these chemicals which do not inhibit the cells growth.
In this study we developed a new tool based on the response of lysosomal enzymes in S. cerevisiae after being exposed with 2 toxic chemicals belong to 2 groups (causing oxidative stress and non-causing oxidative stress agents). Some specific biomarkers were screened and among them, vacuolar protease B (PRB1) - a specific biomarker which had highest up-regulated fold in response with both chemicals, was chosen to fuse with green fluorescent protein (GFP) for the construction of new recombinant yeast. After that, the ability of toxic detection of the new yeast was evaluated by spectrofluorometer in exposing the yeast with pure toxic chemicals and mine water samples.
Materials and Methods
Materials
Sodium meta-arsenite (NaAsO2), tetracycline, aspirin (acetylsalicylic acid) and 2’-7’-dichlorofluoresceindiacetate (Sigma-Aldrich, St. Louis, MO, USA), cadmium nitrate tetrahydrate (CdNO3)2 4H2O (Junsei Chemical, Tokyo, Japan) and Lyso- Tracker Red DND-99 (Molecular Probes, Leiden, Netherlands) were used as the starting materials. Mine water samples were collected from many places and contained various components as mentioned in Table S1.
Yeast Culture, Toxic Treatment and Lysosome Isolation from S. cerevisiae
S. cerevisiae 2805 (MATα pep4::HIS3 prb1-Δ1.6R can1 GAL2 his3-Δ200 ura3-52) (ATCC 208280) was provided by the Korea Research Institute of Bioscience and Biotechnology and grown in yeast extract peptone dextrose (YPD) medium (10 g/ L yeast extract, 20 g/L bacto peptone and 20 g/L glucose) in a 250 mL flask at 30°C and 180 rpm. For investigating the alteration of lysosomal proteins in S. cerevisiae, cells were exposed to the optimal concentrations of tetracycline (111.1 ppm) and Na- AsO2 (1.95 ppm) for 12 hours [6]. Lysosomes were isolated after exposure. After harvesting exponential growth cells, 5 mL of cells were added 25 mL of Tris-SO4 buffer (containing 1 mL of 1 M dithiothreitol [DTT] solution per 100 mL) and then incubated for 15 minutes at 30°C. The mixture was then centrifuged at 3,000 rpm for 5 minutes at 4°C. The supernatant was discarded, and the pellet was collected and suspended in 25 mL of sorbitol K+-phosphate buffer. In the next step, the cells were ultrasonicated the first time at 40 W for 30 minutes (20:10s on/off pulses) and centrifuged at 3,000 rpm (5 minutes, 4°C) to remove the supernatant. The pellet was suspended in 25 mL of breaking buffer (containing 4 mL of 1 M Tris-Cl pH 7.4 and 50 mL of 2,4 M sorbitol in the total volume of 200 mL) and then the mixture was ultrasonicated at 40 W for 20 minutes (10:10s on/off pulses). The mixture was centrifuged at 500×g for 5 minutes before centrifuging the supernatant at 20,000×g for 30 minutes at 4°C. The lysosomes were then collected from the pellet [7].
Protein Extraction for Two-dimensional Electrophoresis
Purified lysosomes were lysed in lysis buffer (20 mM Tris-Cl pH 7.4, 1 mM EDTA pH 8.8, 150 mM NaCl, 1 mM EGTA pH 8.5, 1% v/v Triton X-100), protease inhibitor cocktail, and phenylmethanesulfonyl fluoride (Roche, Indianapolis, IN, USA) with ratio 100:1:1, vigorous agitated on a vortex mixer and flashcooling in ice for 30 minutes. After incubation on ice, the lysates were centrifuged at 13,000 rpm, 4°C for 10 minutes to yield a supernatant. The protein concentration was determined using Bradford assay. The supernatants were stored at -70°C until analyzed by two-dimensional electrophoresis (2-DE) [8].
Two-dimensional Electrophoresis
Proteins were mixed with 350 μL rehydration solution (7 M urea, 2 M Thiourea, 2% 3-[(3-cholamidopropyl) dimethylammonio]- 1-propane-sulfonate (CHAPS) (Sigma-Aldrich), 40 mM Tris (Amersham, Piscataway, NJ, USA), 0.002% bromophenol blue dye (BPB)), 1% DTT (Duchefa Biochemie, Haarlem, Netherlands), and 0.05% immobilized pH gradient (IPG) buffer 3-10 (GE Healthcare, Piscataway, NJ, USA). The first dimension of 2-DE was carried out on a Bio-Rad Protean IEF cell electrophoresis system at 20°C. Linear pH 4-7 IPG gel strips (18 cm, Ready Strip™ IPG strips; Bio-Rad Laboratories, Hercules, CA, USA) were rehydrated overnight by placing the strips gel-side-down in the sample containing rehydration solution in strip holder and covering them with mineral oil (Bio-Rad Laboratories). The samples were loaded with an intracellular protein level of 30 μg for silver staining. Isoelectric focusing was carried out at 20°C with the following setting: 500 V, 6 hours: 1,000 V, 30 minutes: 2,000 V, 30 minutes: 4,000 V, 30 minutes: 8,000 V until 70,000 V and 500 V 12 hours. The IPG gel strips were then equilibrated in an equilibration solution (50 mM Tris-HCl, pH 8.8, 6 M urea, 30% (v/v) glycerol, 2% sodium dodecyl sulfate (SDS), and BPB trace) with 1% DTT for 15 minutes and then 2.5% iodoacetamide for 15 minutes. The equilibrated gel strips were placed in a 12.5% polyacrylamide gel and the second dimensional separation was carried out using a Protean II xi cell system (Bio-Rad, Laboratories). SDS-PAGE was carried out at 200 V, 400 mA for 6 hours at room temperature [9].
Silver Staining
After finishing the SDS-PAGE, the gels were fixed with fixing buffer (50% methanol, 12% acetic acid, 38% distilled water, and 0.00053% formaldehyde) overnight. Then, the gels were washed 2 times with 50% ethanol for 20 minutes before being shaken in the sensitizing solution (0.2 g/L Na2S2O3) for 90 seconds. Next, the gels were washed 3 times with distilled water by shaking at 100 rpm for 20 seconds prior to react with 2 g/L AgNO3 solution during 30 minutes. After that, the stained gels were washed 2 times with distilled water before being developed in a developing solution (60 g/L Na2S2O3, 20 mL of 0.2 g/ L Na2S2O3, 0.00053% formaldehyde). It took around 3-7 minutes to display all protein spots on the gel. The developing step was stopped by transporting the gels into the stopping solution (50% methanol, 12% acetic acid, 38% distilled water) and shaking at 100 rpm for at least 2 hours [9].
Spot Analysis
The staining 2-DE gel was washing 2 times with distilled water at 100 rpm for 15 minutes/time before being scanned by the format of TIFF, 300 dpi. The protein spots in the gel image were detected by Progenesis software (Nonlinear Dynamics, Newcastle, UK). To determine the differently expressed spots, the protein spots from the control sample were used as standard in comparison with the protein spots from the protein samples of S. cerevisiae exposed to NaAsO2 and tetracycline [9].
DNA Isolation, Manipulation and Transformation
Chromosomal DNA was prepared from S. cerevisiae s2805 using a Wizard SV genomic DNA kit (Promega, Madison, WI, USA). Plasmid DNA such as pYES2.0 (Invitrogen, Carlsbad, CA, USA) and pEGFP-C1 (Clontech Laboratories, Palo Alto, CA, USA) were prepared from Escherichia coli cells using an alkaline lysis technique with a QIA spin Miniprep kit (Qiagen, Hilden, Germany). DNA modification, analysis by agarose gel electrophoresis, and ligation were performed using standard procedures [10]. The PCR experiments were carried out using a T Gradient thermocycler (Biometra, Göttingen, Germany), Ex Taq™ DNA polymerase (Takara Bio, Shiga, Japan), and chromosomal DNA as the template. The PCR products were purified using a QIA quick PCR purification kit (Qiagen). The oligonucleotides used for PCR amplification were purchased from Accu Oligo (Bioneer, Daejeon, Korea). Table 1 lists the primers used to introduce the base pair changes. The transformation of E. coli was performed by electroporation with an Electro Cell Manipulator (BTX Technologies, Hawthorne, NY, USA) and yeast transformation was performed by lithium acetate method [11].

Characteristics of S. cerevisiae strains, plasmids and oligonucleotides used in this study
Construction of pYES2::PRB1::GFP in S. cerevisiae
Plasmids used in this study are listed in Table 1. Plasmid pYES2::PRB1::GFP was constructed for expression prb1 with gfp to create a green fluorescent lysosensor. The coding regions prb1 gene was amplified from the chromosomal DNA of S. cerevisiae and gfp gene was amplified from the pEGFP-C1 by PCR using primer pairs in Table 1, respectively. The PCR products were digested with restriction enzymes HindIII/SacI and SacI/ NotI, and then ligated into the plasmid. The yeast transformation was performed by the lithium acetate (LiAc) method [11]. S. cerevisiae s2805 was transformed with pYES2::PRB1::GFP to construct recombinant yeast strain NNT1 (Table 1). The empty vector pYES2.0 was transformed into S. cerevisiae s2805 to construct the control strain NNTM.
Overexpression and Confirmation of the Location of Foreign Proteins in S. cerevisiae
In order to confirm the location of foreign proteins, the expression of PRB1 in S. cerevisiae was examined using the confocal microscope. S. cerevisiae was grown in synthetic defined (SD) medium (6.7 g/L yeast nitrogen base, 2 g/L casamino acid and 20 g/L glucose) and then inducted in synthetic galactose (SG) medium (6.7 g/L yeast nitrogen base, 2 g/L casamino acid and 20 g/L galactose) at 30°C, rinsed with 1× phosphate buffer (PBS), and stained with 100 nM LysoTracker Red DND-99 in PBS for 10 minutes at 30°C. The cells were washed with PBS [12]. Sections were observed under a confocal laser-scanning microscope (LSM 510 META) at the wavelength of 543 nm for lysosomal protein localization, and 488 nm for GFP visualization. The images were generated with a Zeiss LSM image browser [13].
Chemical Exposure and Evaluation by Spectrofluorometer
The recombinant yeast was grown in SD medium and then inducted in SG medium at 30°C before exposing with 4 pure toxic chemicals (heavy metals and pharmaceuticals) and 7 mine water samples for 2 hours. After that, yeast cells were washed with 1×PBS [12]. The fluorescent intensity of green fluorescent protein from S. cerevisiae was assessed by Jasco FP-6500 spectrofluorometer (Jasco, Tokyo, Japan) with the excitation/emission wavelength of 488 nm/509 nm.
Results
Proteomic Analysis of Lysosomes Isolated from S. cerevisiae Exposed to Sodium Meta-arsenite and Tetracycline
The yeast S. cerevisiae was exposed to 2 kind of toxic substances for 12 hours: NaAsO2 which caused oxidative stress, and tetracycline, a non-causing-oxidative stress reagent, which exerted unclearly influence on lysosomes. The results indicated that there were many differently expressed proteins (DEPs) in the case of tetracycline, 12 spots specific for this toxic treatment was found, all of them were up-regulated. Regarding NaAsO2, 43 exclusive spots, of which 34 were up-regulated and 9 were down-regulated.
The 2-DE map showed that all DEPs were located in the region of pH values ranging from 4.0 to 7.0 and protein molecular weights matched the reference spots (Figure 1), which was consistent with the findings from the studies [14,15]. In order to utilize these biomarkers for improving toxic detection capacity, a recombinant yeast contained the biomarker for both toxic compounds were constructed. Based on the results of the differentially expressed lysosomal proteins of S. cerevisiae in response to NaAsO2 and tetracycline (Table 2), we decided to choose vacuolar PRB1, which had the highest up-regulation level under the effect of both toxic agents, for constructing the recombinant yeast targeting non-specific toxic chemicals.

The 2-dimensional electrophoresis map showed the location of differentially expressd proteins in lysosomes of S. cerevisiae in response to (A) no chemical, (B) tetracycline, and (C) NaAsO2. The proteins were separated with pH gradient ranging from 4 to 7 and the molecular weights that were also indicated.
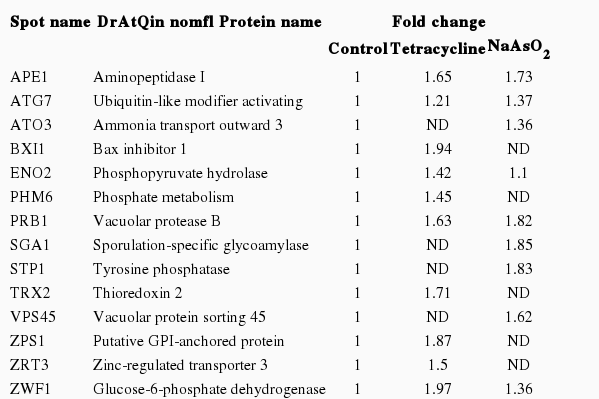
Lists of the differentially expressed lysosomal proteins of S. cerevisiae in response to NaAsO2 and tetracycline.
Construction of the Recombinant Yeast Contained Vacuolar Protease B and Green Fluorescent Protein
PRB1, a single-subunit glycoprotein, is an endoprotease with a pH optimum near neutrality. In the 2-DE work, PRB1 was observed to show the highest up-regulated folds under effects of both tetracycline and NaAsO2 with an increase in the amount of 1.63 and 1.82 folds, respectively. For that reason, PRB1 was expected to be most sensitive to the treatment with these two kinds of chemicals. As a result, PRB1 was chosen as the protein target to construct the recombinant strain containing plasmid pYES2:: PRB1::GFP, named NNT1, which was expected to act on nonspecific toxic agents. This plasmid was sequenced by Bioneer and analyzed using the BLAST tool of National Center of Biotechnology Information to confirm the presence of prb1 and gfp. The analysis resulted in the matching of all insert fragments (data not shown). After that, the recombinant plasmid was transformed into S. cerevisiae by LiAc method.
prb1 was fused with gfp under the control of GAL1 promoter, hence the expression of PRB1 was determined through the visualization of GFP under the confocal microscope. The localization of the fusion protein in lysosome manifested by the merging of green fluorescent of GFP and red fluorescent of lysosomestained LysoTracker. Here, we used the strain NNTM contained mock vector (pYES2) as control. As shown in Figure 2, the merged image displayed a yellow color indicating the combination of green and red color, whereas the control did not show any green or yellow color. This suggested PRB1 was expressed and transported to lysosome.
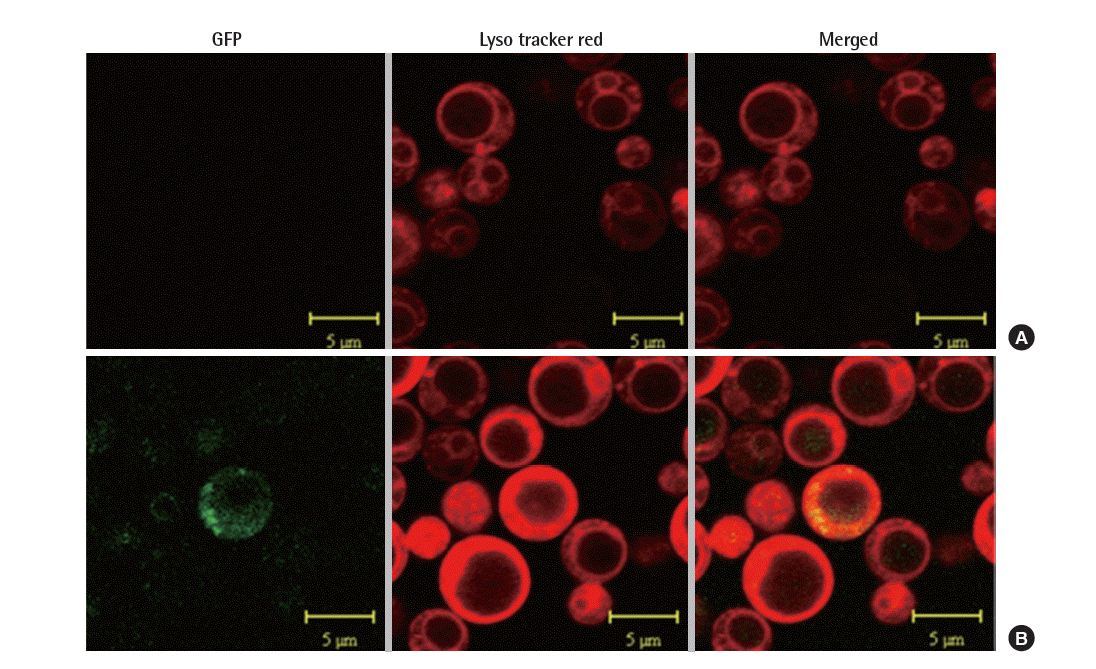
The expression of GFP in lysosome of S. cerevisiae (A) NNTM strain (control strain containing plasmid pYES2), (B) NNT1 strain (recombinant strain containing plasmid pYES2::PRB1::GFP).
Evaluation of the New Strain’s Ability in Toxic Detection in Response with Pure Toxic Chemicals
The yeast was exposed to 4 toxic chemicals at several concentrations. The spectrofluorometer was used to measure the fluorescent intensity due to its sensitivity, simplicity, wide range of wavelengths and time-saving. As shown on Figure 3, each toxic substance had a sub-lethal concentration (cell viability data not shown) at which the fluorescent signal reached the peak (0.65 ppm and 1.54 ppm in case of NaAsO2 and (CdNO3)2 4H2O respectively; and 6.66 ppm, 0.018 ppm with tetracycline and aspirin, correspondingly). In exposing with higher doses, the yeast growth was impacted which provoked the drop of fluorescent intensity. Therefore, this response can be used as the biomarker for the present of this kind of toxic chemicals.
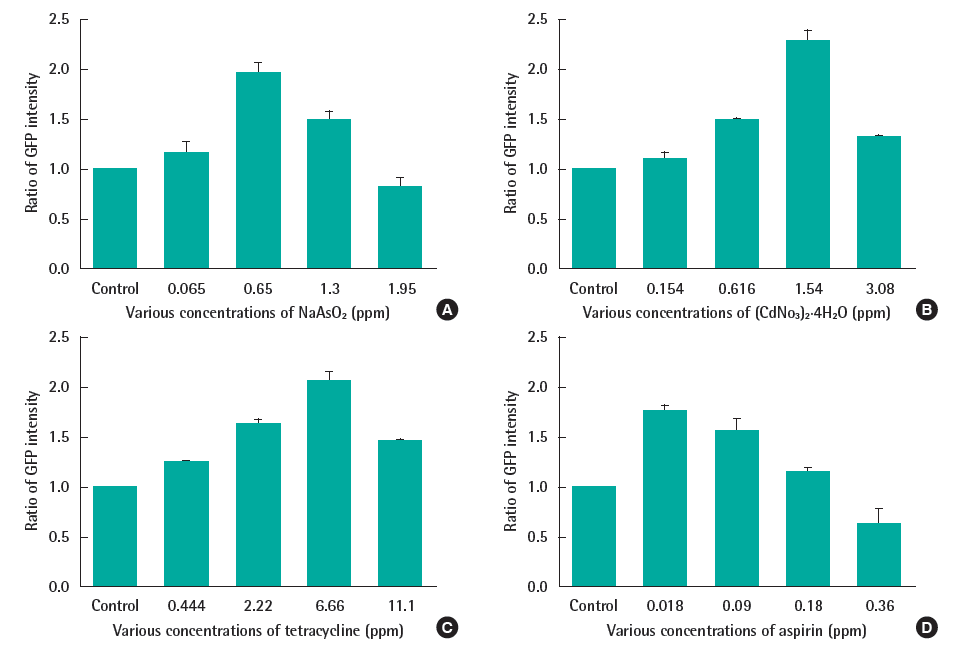
The ratio of GFP intensity in exposing the strain NNT1 (recombinant strain containing plasmid pYES2::PRB1::GFP) with (A) sodium meta-arsenite, (B) (CdNO3)2 4H2O, (C) tetracycline, and (D) aspirin.
Application of Strain NNT1 in Toxic Detection of Mine Water Samples
The yeast was exposed with 7 mine water samples with the informations presented in the Table S1 below. The fluorescent intensity was also assessed by spectrofluorometer at the same condition with previous experiments.
The results were depicted in Figure 4. Regarding water sample 1, the yeast showed higher fluorescent intensity than control at the dilution of 10-3, whereas at higher concentrations, the signal were similar or lower than it. It can be explained by the inhibition of the sample on yeast at high dose which impacted on the protein expression. Similarly, the water sample 3 and 5 caused the overexpression of protein PRB1 at the dilution of 10-5 and 10-4, respectively. Regarding water sample 4, the green signal displayed higher intensity than control at the dilution of 10-2, meanwhile it could not express at lower concentrations.

The ratio of GFP intensity in exposing the strain NNT1 (recombinant strain containing plasmid pYES2::PRB1::GFP) with (A)-(D) water sample 1, 3, 4, and 5. These samples were detectable by the response of GFP; (E)-(G) water sample 2, 6, and 7 which were not detected.
In the case of water sample 2, 6, and 7, the green signal did not indicate any response in exposure with various concentrations of these samples.
Discussion
In order to assess the sensitivity as well as the effect of our system, Daphnia magna - one of the most widely used biosensor for detecting toxic elements currently, was used to compare with. In fact, the initial results obtained in our laboratory seemed to be more promising in comparison with the results obtained with D. magna.
For the comparison of 2 systems, the lethal concentration causing 20% mortality (LC20) of D. magna when exposing with toxic chemicals was used as a reference tool. Regarding heavy metals, it showed that LC20 of D. magna in response with NaAsO2 was 5 ppm, whereas the recombinant yeast’s optimal concentration was 0.65 ppm. However, D. magna was more sensitive in the case of (CdNO3)2 4H2O, with 0.7 ppm caused 20% D. magna mortality while 1.54 ppm generated overexpression of yeast’s PRB1 protein [16]. In addition, the recombinant yeast manifested a clearcut improvement in the case of tetracycline and aspirin. The yeast’s optimal concentrations for response with tetracycline and aspirin are 6.66 ppm and 0.018 ppm, respectively, whereas that of D. magna LC20 was 64 ppm and 295 ppm correspondingly [17].
Regarding the toxic detection with mine water samples, the response varied depend on the components and their doses in each sample. With the sample 1 and 3, the response may be caused by high concentration of alkalinity and NO3 (Table 2) were cytotoxic or the cause of oxidative stress damage. The water sample 4 also contained high dose of alkalinity (67 mg/L) and iron, which was higher than the standard value of iron in South Korea (6.2 mg/L compared to 1-3.5 mg/L standard value). However, the response of PRB1 with iron need further study to be confirmed. The case of sample 5 can be explained by the present of their heavy metal components such as Cd, Co, Cr, Cu, Ni, etc.
In case of undetectable samples (sample 2, 6, and 7), 2 hypothesis could be mentioned. In case of sample 2 and 7, which contained very low level or even no heavy metal and other reactive oxygen species. As a result, the GFP signal was almost observed at extremely low level, which demonstrates that the recombinant yeast has no response to this sample at all. The low signal of GFP was probably not due to the cell death after being treated with these 2 samples since the cell survival remains unaffected (data not shown). In contrast, after being exposed with sample 6, the cell death was observed up to over 70% (data not shown), that is the main reason leading to the very low GFP signal in response to this sample. The explanation for this phenomenon could be because of the high concentration of heavy metal and other reactive oxygen species reactive oxygen species in the ingredient of sample 6. Therefore, further work will be established with lower dose for assessment of PRB1 expression in response with them.
Acknowledgements
This work was supported by the research grant of Chungbuk National University in 2013.
Notes
The authors have no conflicts of interest with material presented in this paper.
References
Supplementary Material
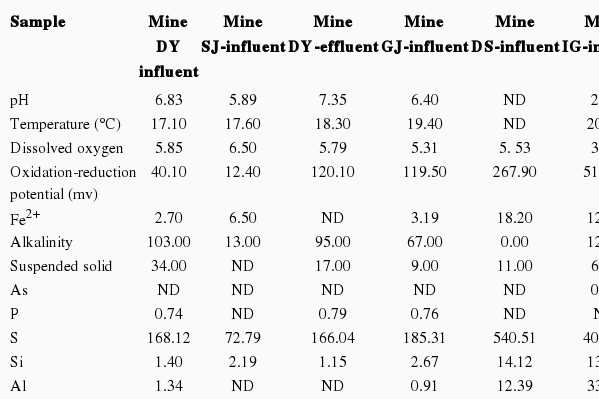
The component concentrations of mine water samples (mg/L)