INTRODUCTION
Quantitative polymerase chain reaction (qPCR) assay has been the flagship of molecular biological methods in numerous scientific fields including soil microbiology. Ever since it was first reported in 1993 [1], it has been by far the most preferred gene quantification method in terms of sensitivity and selectivity [2-5]. Prior to qPCR, DNA extraction is performed via cell lysis such that DNA and other intracellular contents are released from the bacterial cells [6-8].
The adaptation of qPCR for soil microbiology was initially challenging due to the complexity and heterogeneity of soil samples and the fact that soils often vary considerably in physical and chemical properties. Environmental soils often contain the substances that inhibit qPCR, including humic acids, polysaccharides, urea, phenolic compounds and heavy metals. These substances can remain in genomic DNA preparations via coprecipitation during DNA extraction. Subsequently they can cause inhibition to the enzymatic reaction of qPCR [9-12].
In order to circumvent potential inhibition by removing the above-mentioned substances from soil samples, subsequent DNA purification became necessary [10,13-17]. Today’s DNA extraction kit for soil consists of multiple process steps as well as the use of multiple reagents and is suitable even for the most delicate sample.
Unfortunately the DNA purification process is extremely time-consuming (i.e., hours-days) and labor-intensive. In addition, a significant portion of DNA is usually lost during the DNA purification process. In other words, lower recovery yield, degradation, and damage of DNA are common with the extensive DNA purification processes [6,15].
In the advent of big data, Internet-of-Things [18,19], autonomous robots and drones, it appears that soil microbiology can also take on a paradigm shift where we can generate, analyze and share massive amount of information and data. This will have a tremendous impact on our collective perception, understanding and management of soils. In this regard, the DNA purification step during DNA extraction has begun to manifest itself as a painful bottleneck. Perhaps it is time to re-visit the appropriateness and necessity of DNA purification in qPCR assay for soil microbiology.
In this study, we have evaluated the necessity of DNA purification during DNA extraction. The baseline DNA extraction process involved bead beating based lysis [13,15,20-24] followed by centrifugation for the removal of cell debris and soils. The centrifuged lysates were used directly as the template for qPCR assay without additional purification. Its performance was compared with that of conventional DNA extraction kit (with purification). Using Pseudomonas putida seeded sands, various lysis conditions (rpm, duration, and diameter of glass beads) as well as the effect of various amount of inhibitors (humic acids, clay, and magnesium [Mg]) were investigated. Finally, the necessity criteria for purification during DNA extraction with environmental soils were established in relation to humic acids, clay and Mg ion content.
METHODS
Collection and Analysis of Soil Samples
Sand samples washed with water and dried in air were purchased in the form of river-sands from Chungpoong farm in Daejeon, Korea. Three soil samples were collected from Seosan and Gwangyang, Korea. Two rice paddy soil samples from Seosan (soil #1, soil #2) were taken from different following rice fields. One farm soil sample from Gwangyang (soil #3) was taken from the local mixed cultivation farm. Total 100 g of soil samples were collected and prepared by grab sampling method with a single dig per grab for each field. Prior to using them for the experiments, gravels and impurities were first removed by sieving with 1 mm mesh followed by air drying.
The properties of the soil samples were analyzed at several laboratories: soil pH, % organic matter (%OM), and moisture content were analyzed at both Gwangju Institute of Science and Technology (Gwangju, Korea) and Ewha Womans University (Seoul, Korea). The soil texture and the total Mg contents were analyzed at National Instrumentation Center for Environmental Management (Seoul, Korea).
Preparation of P. putida Seeded Sand and Soil Samples
P. putida was chosen as a model bacterium of this study. P. putida is ubiquitous in the aquatic systems and soil habitats and it is more abundant in contaminated soils. Pure strain of P. putida (DSM 8368) was purchased from DSMZ (Braunschweig, Germany). The freeze-dry pellets of P. putida were revived and incubated in the BactoTM tryptic soy broth (Difco Laboratories, Detroit, MI, USA) at ambient temperature using a shaking incubator at 160 rpm. For subsequent experiments, P. putida was harvested at 3 days. The optical density at 600 nm (OD600) of the cells was determined by SpectraMax® M2 microplate reader (Molecular Devices, Sunnyvale, CA, USA).
In order to maintain consistent total cell mass for subsequent experiments, the OD of P. putida in each batch was measured. From the correlation plot between dry cell weight and OD constructed previously [25], final concentration of P. putida used in subsequent experiments was determined to be 0.6 to 0.8 g/L (OD600, 1.8 to 2.1). The harvested P. putida cells were then washed 3 times with 0.01 M, phosphate buffer (PB; pH=7.4) for 3 times and re-suspended in the PB prior to seeding the sand and soil samples. One thousand μL of cells in PB was mixed with 1 g of sand and soil samples (i.e., river sands and environmental soils) in 2 mL vial.
DNA Extraction Without Purification
To present the DNA extraction method without extensive purification, simple bead beating based extraction was employed. Various operational conditions (i.e., beads diameter, beating speed, and duration) were tested with P. putida cell-seeded sands. Five hundred mg of glass beads (Scientific Industries, Bohemia, NY, USA; Glastechnique Mfg, Wertheim, Germany) with various diameters (0.1, 1.0, and 4.0 mm) were used in the experiments. The glass bead mix ratios are as follows: (1) 100% (w/w) of 0.1 mm beads; (2) 50% each of 0.1 and 1.0 mm beads; (3) 50% of 0.1 mm, 30% of 1.0 mm, and 20% of 4.0 mm beads. Each glass bead mix was first added to P. putida cell-seeded sand samples before bead beating at various speeds (1500, 2500, and 3000 rpm) for 0, 1, 2, 3, 5, 7, and 11 minutes. After bead beating, the supernatant was analyzed via the EtBr assay, which is described in the Supplementary Information, and the results were presented as a normalized percent lysis. Centrifugation time was investigated by precipitating non-dissolved lysates (i.e., cell debris) and sands at 5000 rpm for 3, 5, 10, 15, and 20 minutes. Various dilutions of lysates-supernatant (1, 5, and 10 folds) were also tested via qPCR for optimum pretreatment conditions.
Comparison Between Methods With and Without Purification
The operating parameter of the method without purification was determined to be 500 mg of 0.1 mm glass beads at 3000 rpm for 3 minutes. The lysates were further centrifuged at 5000 rpm for 10 minutes and the supernatant was diluted 10 folds with deionized water. Finally, 5 μL of supernatant was subject to the downstream qPCR assay to quantify the gene copies in sand and environmental soil samples. The optimal operating parameters were then used for subsequent comparison experiments.
For comparison, conventional DNA extraction kit (MoBio PowerSoil® DNA isolation kit, Mountain View, CA, USA) was used as a representative to method with purification. The DNA extraction kit was used to extract gDNA from P. putida seeded sand and soil samples in accordance to manufacturer’s protocol. The MoBio kit is based on a bead beating method with 0.7 mm garnet type beads. For the centrifugation, 10 000 rcf (about 10 778 rpm) was used for 30 seconds to 1 minute depending on the steps. At the end of the DNA extraction steps using the kit, the supplied ethylenediaminetetraacetic acid (EDTA)-free elution buffer was used to concentrate gDNA samples. Five μL of the extracted gDNA was then used for the following qPCR assay.
Gene Quantification by Quantitative Polymerase Chain Reaction Assay
Both lysates via the methods with and without purification were used as templates for qPCR assay. The qPCR reaction was performed with Applied Biosystems® StepOne real-time PCR system (Life Technologies, Grand Island, NY, USA). Twenty five μL of qPCR reaction consisted of 1x Applied Biosystems SYBR® Green real-time PCR master mix (Life Technologies), 0.4 μM of both forward primer (5′-GAG ATG CAT ACC ACG TKG GTT GGA-3′) and reverse primer (5′-AGC TGT TGT TCG GGA AGA YWG TGC MGT T-3′), which target polycyclic aromatic hydrocarbon-ring hydroxylating dioxygenases (PAH-RHDα) gene of P. putida [26]. Five μL of each lysate or gDNA (50 ng/μL) was used as the template for each qPCR reaction. The thermal step of qPCR was performed as follows: 95°C for 5 minutes pre-heating step, 40 cycles of 95°C for 30 seconds, 58°C for 30 seconds, 72°C for 30 seconds, extended by 80°C for 10 seconds and 72°C for 7 minutes, and finally followed by a dissociation cycle [26].
The standard gene materials for the calibration curve of gene quantification were obtained in the following procedure: The amplicons (306 bp) of PAH-RHDα were generated by PCR from P. putida culture. The 50 μL PCR assay reaction consisted of 1×TaKaRa PCR buffer (TaKaRa Bio, Shiga, Japan), 1.25 U of TaKaRa Ex Taq® polymerase, 2.5 mM of Mg chloride (MgCl2), 0.2 mM of dNTP and 1 μM of the forward and backward primers (described above). The thermal cycle was in accordance to that reported by Cébron et al. [26], and Looper et al. [27]. The PCR amplicon, purified by ZymocleanTM Gel DNA Recovery kit (Zymo, Irvine, CA, USA), was ligated into the pCR-2.1 TOPO vectors. They were subsequently transformed into the TOP10F’ competent Escherichia coli cells using Invitrogen TOPO® TA Cloning® kit (Life Technologies). The target gene containing plasmids, extracted by QIAprep® Spin Miniprep kit (Qiagen, Venlo, Netherland), were diluted from 102 to 109 gene copies.
Gene copies/g samples (river-sands or environmental soils) were calculated using equation (1) as shown below. This equation was used for both bead beating lysates and gDNA as templates of the qPCR assay.
Where QS is the quantity of target gene obtained from qPCR. D is the dilution factor for the qPCR (in this study, D is 10). Ccell and Vcell refer to bacterial cell concentration (g/L) and volume (L) that was spiked to the sample, respectively. S is the dry soil weight (g) prior to cell spiking. Vbuffer is to the volume (μL) of total lysis buffer and Vtemplate refers to the volume (μL) of template added to each qPCR reaction. In this study, Vbuffer is 1000 μL and Vtemplate is 5 μL, for both methods with and without purification.
Effect of Polymerase Chain Reaction Inhibitors in Soils
The effect of major PCR inhibitors in soils to each treatment was investigated. The inhibitors selected for this study were humic acids, clay, and Mg ions. The following amount of inhibitors was spiked to the P. putida cell seeded sand samples:
Humic acids
Humic acids were obtained from International Humic Substances Society (St. Paul, MN, USA). They were serially diluted with deionized water: 0, 1, 5, 10, 50, 100, 500, 1000, 5000, and 10 000 mg/L. Ten μL of each solution was seeded into 1 g of sand to make 0.0, 0.01, 0.05, 0.1, 0.5, 1, 5, 10, 50, and 100 μg/g sand as a final concentration.
Clay
As a representative of clay, kaolinite (Junsei, Tokyo, Japan) was used. The kaolinite powder was mixed with sand samples to obtain the final composition of 0, 10, 15, 20, 32.5, 55, 60, 70, 80, and 100% (w/w). One g of clay-sand mixture was used for the experiment.
Magnesium
MgCl2 was purchased from Daejung (Seongnam, Korea). Two M of stock solution was prepared by dissolving 4.1825 g of MgCl2 hexahydrate in 10 mL of deionized water. The filter-sterilized stock solution with Millex® syringe filter was then serially diluted with deionized water. The final concentrations of diluted solution were 0.0, 0.004, 0.02, 0.04, 0.2, 0.4, 0.8, 1.2, 1.6, and 2.0 M. One hundred μL of each reagent was spiked into 1 g of sand sample. This means the sand samples would contain 0.001, 0.005, 0.01, 0.05, 0.1, 0.2, 0.3, 0.4, and 0.5% (w/w) Mg ion content, respectively.
Subsequently, three inhibitors laden sands were subject to both methods with and without purification. Five μL of the lysates or gDNA (triplicate sample) were then used for subsequent qPCR assay.
RESULTS
DNA Extraction With and Without Purification
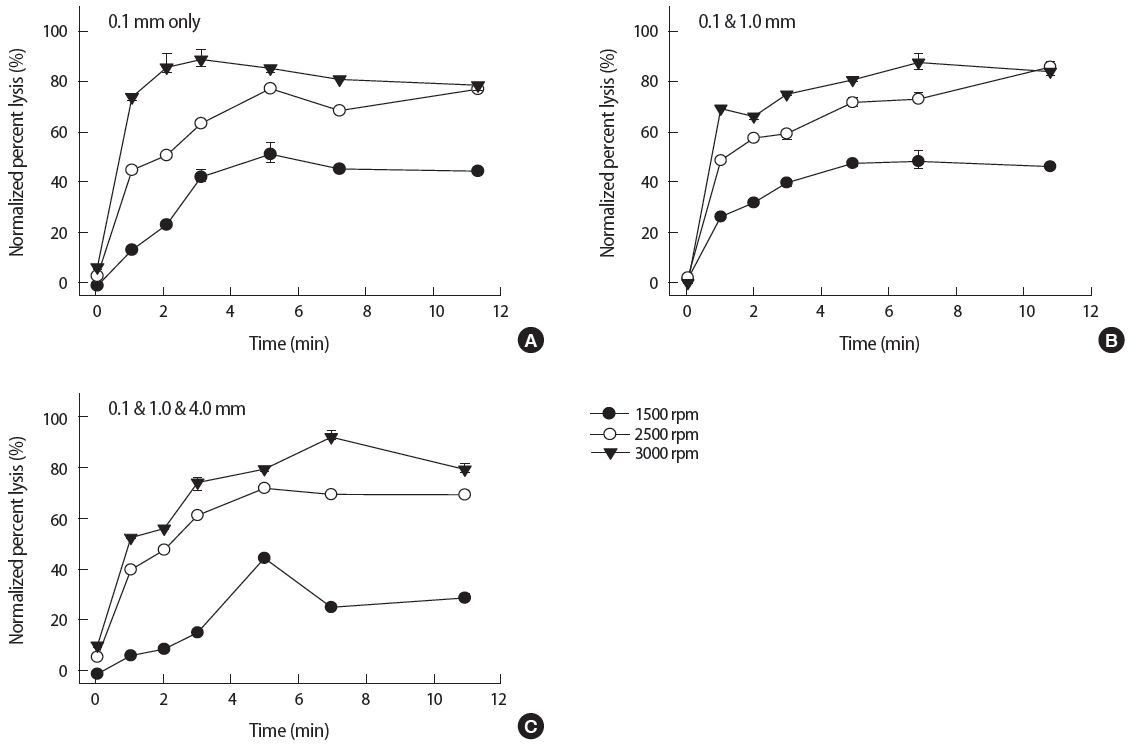
Figure 1 shows the normalized percent lysis (%) of the baseline method without purification. The lysis was shown as a function of different beating speed, duration, and bead diameter. The overall trend of lysis over time indicated that the normalized % lysis drastically increased at the initial 1 minute and it slowly increased afterwards. The maximum speed at 3000 rpm (reversed solid triangles) showed the highest lysis (about 90%) for all three bead combinations. As expected, the lower bead beating speed resulted in decreased yields compared to the 3000 rpm setting. When the samples were treated at 2500 rpm (open circles) for 3 minutes, about 60% lysis was obtained. Bead beating at 1500 rpm (solid circles) for 3 minutes showed about 40% lysis.
Pertaining to the effect of different bead diameters, 0.1 mm beads only (Figure 1A) showed about 90% lysis within 3 minutes at 3000 rpm. As compared to 0.1 mm beads only, two other mix ratio of 0.1 and 1.0 mm and 0.1, 1.0, and 4.0 mm (Figure 1B and 1C, respectively) required at least 7 minutes to reach the same lysis (about 90%) at the same speed. All three cases at 2500 rpm showed the similar lysing performance over time regardless of the bead diameter. However, at 1500 rpm, the mixture of 0.1, 1.0, and 4.0 mm beads (Figure 1C) showed the least amount of lysis (about 20%) at 3 minutes. Overall results indicated the optimal parameters were 3000 rpm for 3 minutes with 0.1 mm beads only.
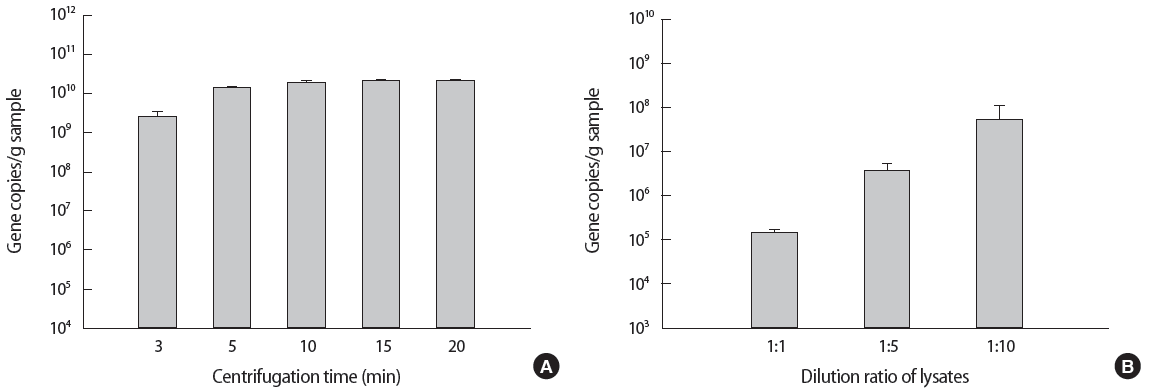
The optimum centrifugation time and dilution ratio of lysates were determined in accordance to the qPCR result. As shown in Figure 2A, the gene copies/g sample increased as the centrifugation time increased. At 10 minutes, the gene copies/g sample appeared to reach its maximum. Subsequent increase in centrifugation time (15 and 20 minutes) did not yield further increase in gene copies/g sample (p=0.38, analysis of variance [ANOVA] test, no significant difference among 10, 15, and 20 minutes). Therefore 10 minutes of centrifugation was determined to be optimal.
Dilution ratio of lysates was also investigated and presented in Figure 2B. When the supernatant was diluted to 10 folds, maximum gene copies were obtained by qPCR. Therefore the supernatant was 10 folds diluted subsequent to 10 minutes of centrifugation.
Comparison of Gene Quantification Between Methods With and Without Purification
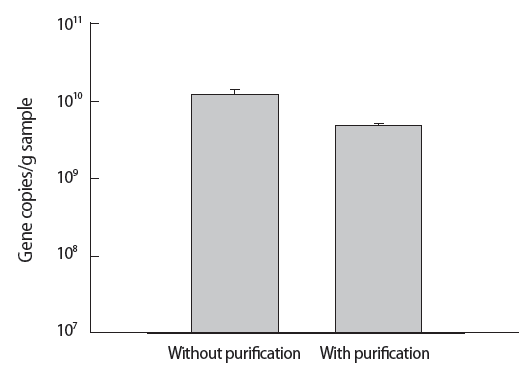
The PAH-RHDα gene quantification by qPCR was performed to compare DNA extraction with and without purification (Figure 3). Gene copies/g sand sample indicated the method without purification (1.2×1010) showed higher value than the method with purification (4.8×109). The difference of gene copies/g sample was statistically significant (p=0.009, t-test) between the method with and without purification.
Effect of Polymerase Chain Reaction Inhibitors
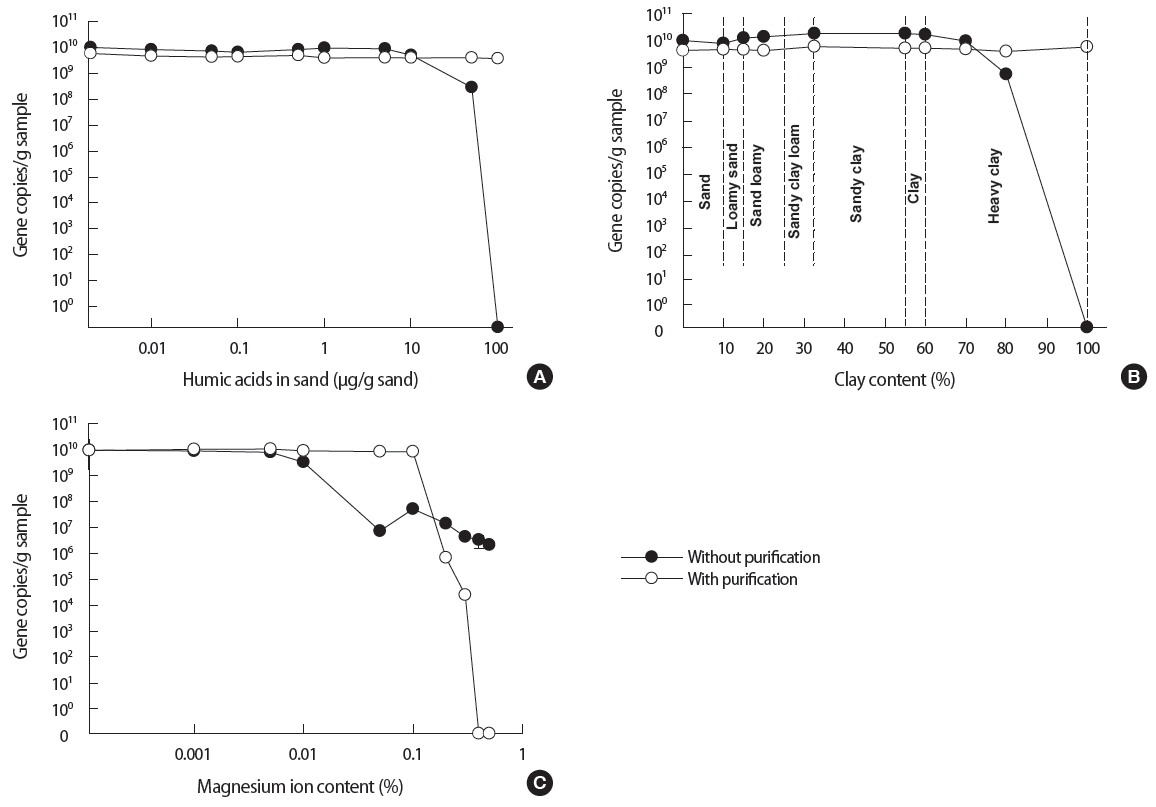
The effect of selected inhibitors (humic acids, clay and Mg ion) to the each treatment was investigated using sand samples. The comparison result was shown for the DNA extraction with and without purification (Figure 4). As shown in Figure 4A, gene copies/g sample by the DNA extraction without purification were consistent and slightly higher than that by the conventional DNA extraction kit. This trend continued till the humic acids reached a concentration of 10 μg/g sand by showing average values in this range as 8.3×109 gene copies/g sample for without purification and 4.7×109 gene copies/g sample with purification, respectively. At this point, the gene copies/g sample by the method without purification began to decrease. At humic acids concentration of 50 μg/g, the gene copies/g sample by the method without purification have decreased by about one order of magnitude. At humic acids concentration of 100 μg/g, the lysates from the method without purification ceased to produce meaningful results on the qPCR assay.
In the case of clay as an inhibitor (Figure 4B), both methods have shown approximately 4.8×109 and 1.3×1010 gene copies/g sample consistently for clay content of up to 70%. However at higher clay content (80 to 100%), the performance of the method without purification began to reduce significantly.
The effect of Mg ion as an inhibitor is interesting (Figure 4C). At the range of 0.001 to 0.01% Mg ion content, both methods achieved approximately 109 to 1010 gene copies/g sample. At the Mg2+ content of 0.01 to 0.05%, the gene copies from the method without purification decreased by 2 to 3 orders of magnitude before stabilizing at approximately 107 gene copies. The method with purification was able to maintain consistent gene copies up to 0.1% of Mg ion content. However at Mg ion content of more than 0.1%, the performance of the method with purification began to reduce significantly.
Environmental Soil Samples
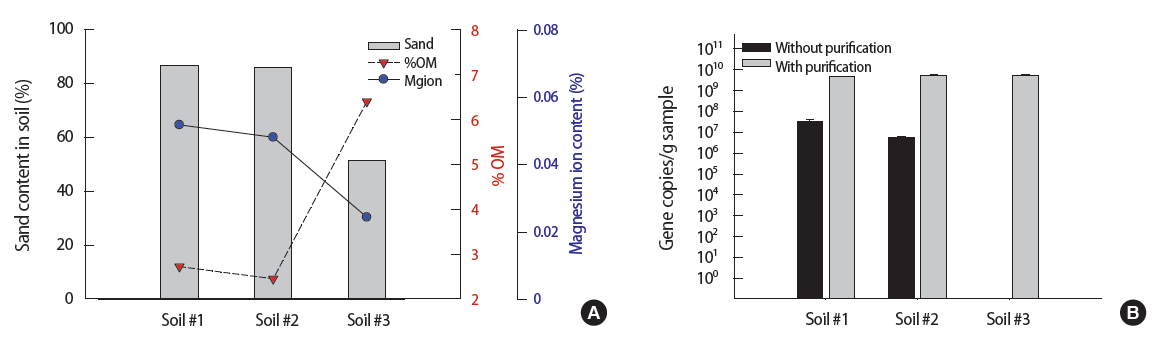
The key soil properties of three farm soil samples were described in Table 1. Two different rice paddy soil samples from Seosan (soil #1 and #2) showed the similar physico-chemical characteristics. Both soil #1 and #2 had around 86% sand content, which is classified as a loamy sand. The %OM (w/w) values of soil #1 and #2 were relatively low (2.5 to 2.7%). Compared to soil #1 and #2, the farm soil sample from Gwangyang area (soil #3) has shown the lower sand content (51.5%) and higher %OM (6.4%). Soil #3 is close to sandy clay or loam based on 28. US Department of Agriculture textural triangle [28]. In terms of total Mg content, soil #1 and #2 showed about 2 times higher (10 352 mg/kg and 9614 mg/kg, respectively) than soil #3 (4861 mg/kg). Based on the study by Mengel et al. [29], Mg ion in general constitutes 5% of total Mg content. Using this conversion ratio, the total Mg concentration in soil samples were converted to Mg ion content (%), where soils #1, #2, and #3 contained 0.052, 0.048, and 0.024% of Mg ion, respectively.
Based on the soil properties described in Figure 5A, the soil #3 contains lower sands (51.5%), lower Mg ion (0.024%), and higher %OM (6.4%) as compared to soil #1 and soil #2 (0.052%, 0.048% for Mg ion and 2.7%, 2.5% for %OM, respectively). The quantitative results of PAH-RHDα gene for three seeded soil samples via qPCR assay are shown in Figure 5B. As expected, the method with purification (gray-colored bar) showed similar values for all three soil samples (4.9×109, 5.5×109, 5.3×109 gene copies/g sample for soil #1, #2, and #3, respectively) with no significant difference (p=0.46, 95% confidence interval, ANOVA test). As for the method without purification, the gene quantities of soil #1 and #2 were 3.4×107, 5.5×106 gene copies/g sample. Two to three orders of magnitude was reduced when the method without purification was used (p=0.0008, 0.008 for #1 and #2, respectively, being significantly different between with and without purification). It indicated the slight inhibition occurs during qPCR. In the case of soil #3, it showed almost zero gene copies/g sample, indicating the method without purification is not suitable for soil #3.
DISCUSSION
Allowable Range of Inhibitors in Soil Samples for the Method Without Purification
In order for us to investigate the necessity of purification during DNA extraction, the effect of PCR inhibitors existing in soil samples has to be examined for both methods with and without purification. Therefore the corresponding allowable range of inhibitors in soil samples can be established for the method without purification. This is especially critical when the relatively ‘dirty’ environmental samples are to be subjected to analysis.
Humic acids is one of the most common inhibitors existing in environmental soils. Several studies have shown that the humic acids can inhibit the PCR at the amplification steps or have adverse effects on the DNA extraction via sequestration [12,30]. Based on the result presented in Figure 4A, the method without purification can be used as an alternative to the method with purification when humic acids concentration is less than 10 μg/g sample.
As shown in Figure 4B, the method without purification is only compromised at clay contents of 70% or higher. This is an interesting observation because clay is anticipated to play a significant inhibitory role. The lysed DNA can be adsorbed into the clay surface due to the charge characteristics [17]. Once the lysed DNA is adsorbed into clay, it would not be entirely available for the downstream qPCR amplification. According to the review by Yu et al. [31], the clay-DNA adsorption involves ligand exchange, cation bridge, electrostatic force, and hydrogen bonding. Among the various interactions, ligand exchange and electrostatic force may be the major mechanisms that hinder PCR amplification with the lysed DNA. First of all, the phosphate groups at the end of DNA fragments are directly bound to the hydroxyl groups on the edge of clay minerals. It refers to the ligand exchange. Secondly, inorganic cations in environmental samples such as Mg ion, calcium ion and potassium ion tend to form cation bridges with the clay particles. It makes the clay surface positive. Therefore the negatively charged (by phosphate groups) DNA molecules are electrostatically attracted to the positively charged clay surface.
Yet as shown in Figure 4B, clay played a surprisingly minor role in inhibition during the employment of the method without purification. This could be due to the use of PB (pH=7.4) in the method without purification. The buffer maintains the pH as well as provides a number of phosphate ions. It was known that the pH of soil and hence that of the PB is an influential factor to the adsorption of DNA into the clay [17]. As the pH increases, the positively charged clay particles become negatively charged via deprotonation of AlOH to AlO-. Subsequently, the adsorption of DNA into clay decreases as the electrostatic repulsion increases between the DNA and clay [32-35]. The pH of the buffer used in this study was 7.4 and it appeared sufficiently high to prevent the adsorption between the lysed DNA and clay particles. Moreover, saturated phosphate ions provided by the buffer solution are likely to compete with DNA phosphate groups to increase electrostatic repulsion [31, 36,37].
Overall this is a pleasant discovery because soil samples with clay contents of 70% or higher is deemed ‘heavy clay’ and is not environmentally relevant. In other words, most of the environmental soil samples will have clay content lower than 70%. Hence we can conclude that the purification processes may not necessary for most environmental soil samples as far as clay contents are concerned.
As mentioned earlier, Mg ion is found ubiquitously in environmental soils. The Mg ion is a co-factor of polymerase therefore it can improve enzymatic activity by affecting the primer annealing and DNA melting temperature [38]. However, excessive Mg ion will result in lower performance during PCR reaction. As mentioned in the review by Shamsi and Kraatz [39], multivalent cations such as Mg ion tend to bind with nucleic acids more tightly than other monovalent cations.
Once DNA is combined with the Mg ions, the denaturation of dsDNA can become incomplete during PCR amplification [40, 41]. Therefore the conventional DNA extraction kit often contains EDTA reagents to capture and separate metal ions from the extracted DNA. However as shown in Figure 4C, even the conventional DNA extraction kit is unable to prevent inhibition at Mg content exceeding 0.1%.
In comparison, the method without purification was only showed slight inhibition (gene copies up to 107 copies) when the method with purification showed complete inhibition. This surprising observation may be explained by the use of phosphate based lysis buffer in the method without purification. The negatively charged phosphate groups in the buffer might have competed with the lysed DNA in their interaction with Mg ion. In other words, the phosphate groups literally reduced the amount of Mg ion by forming Mg-phosphate complexes. This means less amount of lysed DNA would be lost to the formation of Mg- DNA complexes.
Nonetheless more work is needed to elucidate other possible reactions with various inhibitors and the lysis buffer. Although some studies observed that there is no marked difference on the DNA yield as a result of various type of lysis buffer [21,42], the results in this study suggest that the lysis buffer can be an important factor that influences the degree of inhibition (or mitigation) by common environmental inhibitors.
Environmental Soil Samples
Using the method without purification (Figure 5B), soil #3 (%OM about 6.4%) yielded almost no gene copies as compared to soils #1 and #2 (%OM about 2.5%). This is consistent with the results shown in Figure 4A where higher humic acids content (indicated by %OM) implied more inhibition. In this case, it is likely that soil #3 has humic acids content of at least 100 μg/g sample.
Comparing the method without purification with the method with purification for soils #1 and #2, it is apparent that there is some degree of inhibition in the method without purification. Since the Mg ion content of both soils #1 and #2 is about 0.05%, it implied from Figure 4C that the method without purification should experience inhibition in the form of reduced gene copies by 2 to 3 orders of magnitude. This is consistent with Figure 5 where the method without purification also experienced similar degree of inhibition in the form of reduced gene copies by 2 to 3 orders of magnitude.
At this juncture, it is apparent that the method without purification is suitable for soil samples that have less than 10 μg/g of humic acids, less than 70% clay content and less than 0.01% Mg ion content. For such soil samples, the intensive purification that is commonly employed in the conventional DNA extraction kit may not be a viable strategy for its DNA preparation. In order to extend the DNA preparation without purification for soil samples that have higher humic acids, clay content or Mg ion content, the effect of inhibition needs to be taken into account to arrive at an appropriate compensation algorithm. Fortunately, it appears that the degree of inhibition may be predicted based on the soil property measurements. Hence it is possible that we can develop an appropriate compensation algorithm in the future in order to bypass DNA purifications for as many types of soils as possible. In the other hand, we aware that linking soil properties to the necessity of DNA purification requires extensive validations with a variety of natural soil testing and the elucidation of the interaction effects.